

2023
D. Gryko; A. Szumna; A. Riina; T. Soós
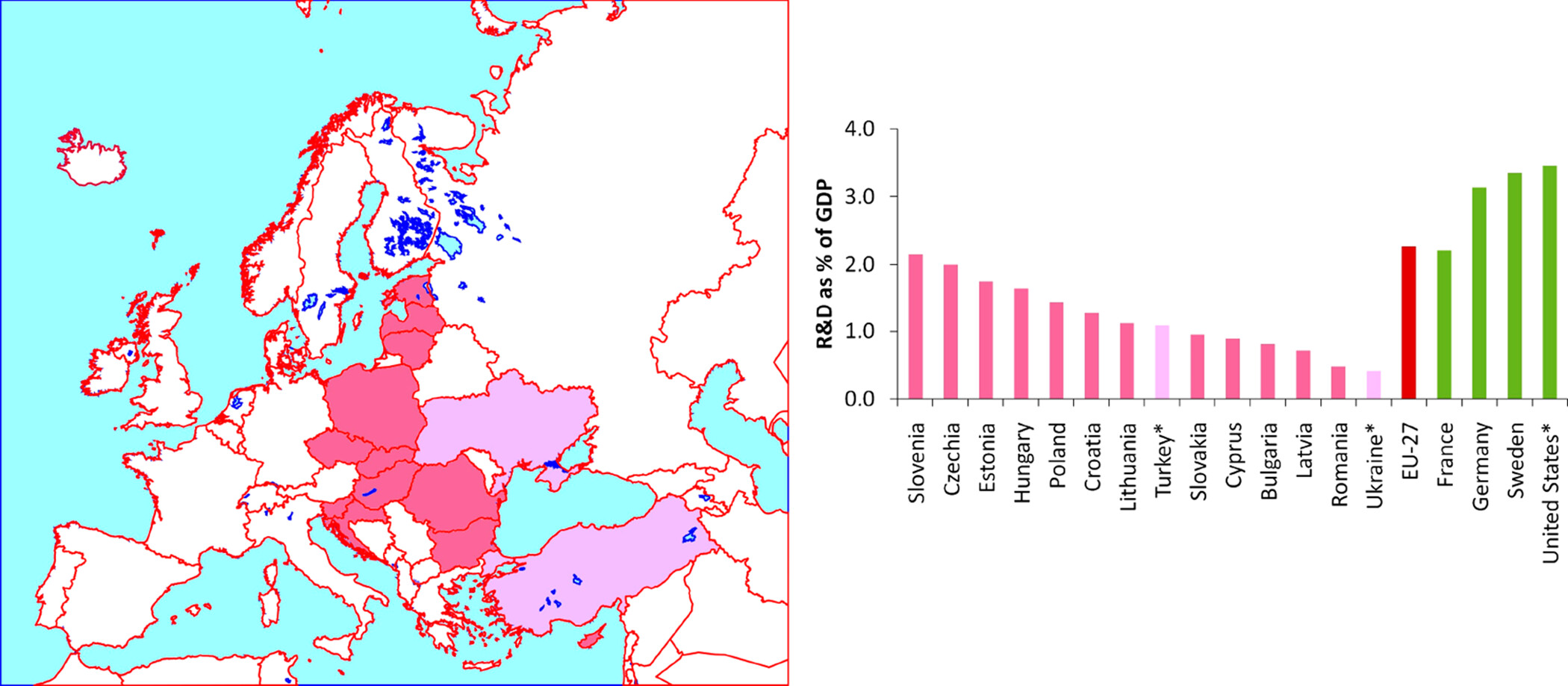
ChemEASTry Europe, the Status of the Chemical Sciences in a Growing Region Journal Article
In: Organic Letters, vol. 25, iss. 34, pp. 6237 - 6239, 2023.
Links | Tags:
@article{nokey,
title = {ChemEASTry Europe, the Status of the Chemical Sciences in a Growing Region},
author = {D. Gryko and A. Szumna and A. Riina and T. Soós},
doi = {10.1021/acs.orglett.3c02621},
year = {2023},
date = {2023-09-01},
urldate = {2023-09-01},
journal = {Organic Letters},
volume = {25},
issue = {34},
pages = {6237 - 6239},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
G. Sztanó; Z. Dobi; T. Soós
Strain and Complexity, Passerini and Ugi Reactions of Four-Membered Heterocycles and Further Elaboration of TOSMIC Product Journal Article
In: ChemistryOpen, vol. 12, iss. 8, pp. e202200083, 2023.
@article{nokey,
title = {Strain and Complexity, Passerini and Ugi Reactions of Four-Membered Heterocycles and Further Elaboration of TOSMIC Product},
author = {G. Sztanó and Z. Dobi and T. Soós},
doi = {10.1002/open.202200083},
year = {2023},
date = {2023-08-07},
urldate = {2023-08-07},
journal = {ChemistryOpen},
volume = {12},
issue = {8},
pages = {e202200083},
abstract = {Straightforward and general Passerini and Ugi procedures have been developed to incorporate four-membered heterocycles into highly functionalized scaffolds. Additionally, toslymethyl isocyanide (TosMIC)-derived Ugi adducts have been prepared, showcasing the prospect of the multicomponent reaction.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

P. Angyal; K. Hegedüs; B. B. Mészáros; J. Daru; Á. Dudás; A. R. Galambos; N. Essmat; M. Al-Khrasani; Sz. Varga; T. Soós
Total Synthesis and Structural Plasticity of Kratom Pseudoindoxyl Metabolites Journal Article
In: Angewandte Chemie - International Edition, vol. 62, iss. 35, pp. e202303700, 2023.
@article{nokey,
title = {Total Synthesis and Structural Plasticity of Kratom Pseudoindoxyl Metabolites},
author = {P. Angyal and K. Hegedüs and B. B. Mészáros and J. Daru and Á. Dudás and A. R. Galambos and N. Essmat and M. Al-Khrasani and Sz. Varga and T. Soós},
doi = {10.1002/anie.202303700},
year = {2023},
date = {2023-06-18},
urldate = {2023-06-18},
journal = {Angewandte Chemie - International Edition},
volume = {62},
issue = {35},
pages = {e202303700},
abstract = {Mitragynine pseudoindoxyl, a kratom metabolite, has attracted increasing attention due to its favorable side effect profile compared to conventional opioids. Herein, we describe the first enantioselective and scalable total synthesis of this natural product in addition to its epimeric congener, speciogynine pseudoindoxyl. The characteristic spiro-5-5-6 tricyclic system of these alkaloids is formed via a protecting group-free cascade relay process in which oxidized tryptamine and secologanin analogs are used. Furthermore, we uncovered that mitragynine pseudoindoxyl acts not as a single molecular entity but as a dynamic ensemble of stereoisomers in protic environments, thus, it exhibits structural plasticity in biological systems. Accordingly, these synthetic, structural, and biological studies provide a basis for the planned design of mitragynine pseudoindoxyl analogs, which can guide the development of next-generation analgesics.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

B. Kőnig; G. Sztanó; T. Holczbauer; T. Soós
Syntheses of 2- and 3-Substituted Morpholine Congeners via Ring Opening of 2-Tosyl-1,2-Oxazetidine Journal Article
In: Journal of Organic Chemistry, vol. 88, iss. 9, pp. 6182 - 6191, 2023.
@article{nokey,
title = {Syntheses of 2- and 3-Substituted Morpholine Congeners via Ring Opening of 2-Tosyl-1,2-Oxazetidine},
author = {B. Kőnig and G. Sztanó and T. Holczbauer and T. Soós },
doi = {10.1021/acs.joc.3c00207},
year = {2023},
date = {2023-04-26},
urldate = {2023-04-26},
journal = {Journal of Organic Chemistry},
volume = {88},
issue = {9},
pages = {6182 - 6191},
abstract = {Diastereoselective and diastereoconvergent syntheses of 2- and 3-substituted morpholine congeners are reported. Starting from tosyl-oxazatedine 1 and α-formyl carboxylates 2, base catalysis is utilized to yield morpholine hemiaminals. Their further synthetic elaborations allowed the concise constructions of conformationally rigid morpholines. The observed diastereoselectivities and the unusual diastereoconvergence in the photoredox radical processes seem to be the direct consequence of the avoidance of pseudo A1,3 strain between the C-3 substituent and the N-tosyl group and the anomeric effect of oxygen atoms.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

K. Albitz; D. Csókás; Z. Dobi; I. Pápai; T. Soós
Late-Stage Formal Double C–H Oxidation of Prenylated Molecules to Alkylidene Oxetanes and Azetidines by Strain-Enabled Cross-Metathesis Journal Article
In: Angewandte Chemie - International Edition, vol. 62, no. 13, pp. e202216879, 2023.
@article{nokey,
title = {Late-Stage Formal Double C–H Oxidation of Prenylated Molecules to Alkylidene Oxetanes and Azetidines by Strain-Enabled Cross-Metathesis},
author = {K. Albitz and D. Csókás and Z. Dobi and I. Pápai and T. Soós},
doi = {10.1002/anie.202216879},
year = {2023},
date = {2023-01-11},
urldate = {2023-01-11},
journal = {Angewandte Chemie - International Edition},
volume = {62},
number = {13},
pages = {e202216879},
abstract = {Prenylation is a ubiquitous late-stage modification in nature that often confers significantly improved bioactivity for secondary metabolites. While this lipophilic modification renders enhanced potency, the lipophilic tag(s) can diminish bioavailability and adversely alter drug transportation and metabolism. Thus, a functional group tolerant, mild, and selective late-stage C–H functionalization of prenyl tags would present a great potential in drug discovery programs but could also impact other fields, such as agrochemistry and chemical biology. Herein we report an exocyclic strain driven cross-metathesis reaction of prenyl tags, a formal double C–H oxidation protocol, that can be used for the selective late-stage derivatization of prenylated compounds and natural products. This methodology avoids the need of pre-functionalization of target molecules and affords an easy access to an unprecedented library of oxo- and aza-prenylated complex molecules. Thus, in a broader context, this methodology extends late-stage functionalization beyond that available to nature.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

P. Angyal; A. M. Kotschy; Á. Dudás; Sz. Varga; T. Soós
Intertwining Olefin Thianthrenation with Kornblum/Ganem Oxidations: Ene-type Oxidation to Furnish α,β-Unsaturated Carbonyls Journal Article
In: Angewandte Chemie - International Edition, vol. 62, no. 2, pp. e202214096, 2023.
@article{nokey,
title = {Intertwining Olefin Thianthrenation with Kornblum/Ganem Oxidations: Ene-type Oxidation to Furnish α,β-Unsaturated Carbonyls},
author = {P. Angyal and A. M. Kotschy and Á. Dudás and Sz. Varga and T. Soós},
url = {https://www.scopus.com/record/display.uri?eid=2-s2.0-85143289733&origin=resultslist&sort=plf-f&src=s&st1=10.1002%2fanie.202214096&sid=2d94cac761be987eb427fb393388597f&sot=b&sdt=b&sl=27&s=DOI%2810.1002%2fanie.202214096%29&relpos=0&citeCnt=0&searchTerm=},
doi = {10.1002/anie.202214096},
year = {2023},
date = {2023-01-09},
urldate = {2023-01-09},
journal = {Angewandte Chemie - International Edition},
volume = {62},
number = {2},
pages = {e202214096},
abstract = {A widely applicable, practical, and scalable synthetic method for efficient ene-type double oxidation of alkenes is reported via a two-step alkenyl thianthrenium umpolung/Kornblum-Ganem oxidation strategy. This chemo- and stereoselective procedure allows easy access to various α,β-unsaturated carbonyls that may be otherwise difficult or cumbersome to synthesize by conventional methods. For α-olefins, this metal-free transformation can be tuned according to synthetic needs to produce either the elusive (Z)-unsaturated aldehydes or their (E) counterparts. Moreover, this strategy has enabled streamlined synthesis of distinct butadienyl pheromones and kairomones},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

2022
V. F. S. Pape; R. Palkó; Sz. Tóth; M. J. Szabó; J. Sessler; Gy. Dormán; É. A. Enyedy; T. Soós; I. Szatmári; G. Szakács
Structure–Activity Relationships of 8-Hydroxyquinoline-Derived Mannich Bases with Tertiary Amines Targeting Multidrug-Resistant Cancer Journal Article
In: Journal of Medicinal Chemistry, vol. 65, no. 11, pp. 7729-7745, 2022.
@article{nokey,
title = {Structure–Activity Relationships of 8-Hydroxyquinoline-Derived Mannich Bases with Tertiary Amines Targeting Multidrug-Resistant Cancer},
author = {V. F. S. Pape and R. Palkó and Sz. Tóth and M. J. Szabó and J. Sessler and Gy. Dormán and É. A. Enyedy and T. Soós and I. Szatmári and G. Szakács},
url = {https://www.scopus.com/record/display.uri?eid=2-s2.0-85131772546&origin=resultslist&sort=plf-f&src=s&st1=10.1021%2facs.jmedchem.2c00076&sid=d86b487408ff007a7b4169c88f878544&sot=b&sdt=b&sl=33&s=DOI%2810.1021%2facs.jmedchem.2c00076%29&relpos=0&citeCnt=3&searchTerm=},
doi = {10.1021/acs.jmedchem.2c00076},
year = {2022},
date = {2022-05-25},
urldate = {2022-05-25},
journal = {Journal of Medicinal Chemistry},
volume = {65},
number = {11},
pages = {7729-7745},
abstract = {A recently proposed strategy to overcome multidrug resistance (MDR) in cancer is to target the collateral sensitivity of otherwise resistant cells. We designed a library of 120 compounds to explore the chemical space around previously identified 8-hydroxyquinoline-derived Mannich bases with robust MDR-selective toxicity. We included compounds to study the effect of halogen and alkoxymethyl substitutions in R5 in combination with different Mannich bases in R7, a shift of the Mannich base from R7 to R5, as well as the introduction of an aromatic moiety. Cytotoxicity tests performed on a panel of parental and MDR cells highlight a strong influence of experimentally determined pKa values of the donor atom moieties, indicating that protonation and metal chelation are important factors modulating the MDR-selective anticancer activity of the studied compounds. Our results identify structural requirements increasing MDR-selective anticancer activity, providing guidelines for the development of more effective anticancer chelators targeting MDR cancer.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

2020
S. Varga; P. Angyal; G. Martin; O. Egyed; T. Holczbauer; T. Soós
Total Syntheses of (−)-Minovincine and (−)-Aspidofractinine through a Sequence of Cascade Reactions Journal Article
In: Angewandte Chemie - International Edition, vol. 59, no. 32, pp. 13547-13551, 2020.
@article{Varga202013547,
title = {Total Syntheses of (−)-Minovincine and (−)-Aspidofractinine through a Sequence of Cascade Reactions},
author = {S. Varga and P. Angyal and G. Martin and O. Egyed and T. Holczbauer and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85085900974&doi=10.1002%2fanie.202004769&partnerID=40&md5=63fae7eac53f87f7c15894d345695a7d},
doi = {10.1002/anie.202004769},
year = {2020},
date = {2020-01-01},
urldate = {2020-01-01},
journal = {Angewandte Chemie - International Edition},
volume = {59},
number = {32},
pages = {13547-13551},
abstract = {We report 8-step syntheses of (−)-minovincine and (−)-aspidofractinine using easily available and inexpensive reagents and catalyst. A key element of the strategy was the utilization of a sequence of cascade reactions to rapidly construct the penta- and hexacyclic frameworks. These cascade transformations included organocatalytic Michael-aldol condensation, a multistep anionic Michael-SN2 cascade reaction, and Mannich reaction interrupted Fischer indolization. To streamline the synthetic routes, we also investigated the deliberate use of steric effect to secure various chemo- and regioselective transformations. © 2020 The Authors. Published by Wiley-VCH Verlag GmbH & Co. KGaA.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

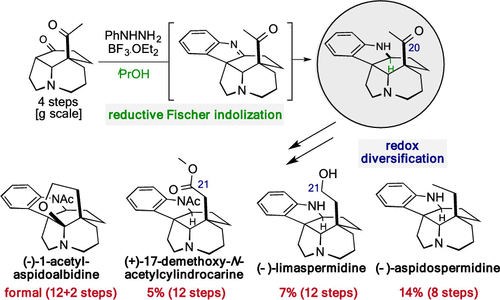
G. Martin; P. Angyal; O. Egyed; S. Varga; T. Soós
Total Syntheses of Dihydroindole Aspidosperma Alkaloids: Reductive Interrupted Fischer Indolization Followed by Redox Diversification Journal Article
In: Organic Letters, vol. 22, no. 12, pp. 4675-4679, 2020.
@article{Martin20204675,
title = {Total Syntheses of Dihydroindole Aspidosperma Alkaloids: Reductive Interrupted Fischer Indolization Followed by Redox Diversification},
author = {G. Martin and P. Angyal and O. Egyed and S. Varga and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85086572635&doi=10.1021%2facs.orglett.0c01472&partnerID=40&md5=2ba602067c507b05209bbe2ff847a8f7},
doi = {10.1021/acs.orglett.0c01472},
year = {2020},
date = {2020-01-01},
urldate = {2020-01-01},
journal = {Organic Letters},
volume = {22},
number = {12},
pages = {4675-4679},
abstract = {We report a novel reductive interrupted Fischer indolization process for the concise assembly of the 20-oxoaspidospermidine framework. This rapid complexity generating route paves the way toward various dihydroindole Aspidosperma alkaloids with different C-5 side chain redox patterns. The end-game redox modulations were accomplished by modified Wolff-Kishner reaction and photo-Wolff rearrangement, enabling the total synthesis of (-)-aspidospermidine, (-)-limaspermidine, and (+)-17-demethoxy-N-acetylcylindrocarine and the formal total synthesis of (-)-1-acetylaspidoalbidine. Copyright © 2020 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2019
P. Spránitz; P. Soregi; B. B. Botlik; M. Berta; T. Soós
Organocatalytic Desymmetrisation of Fittig's Lactones: Deuterium as a Reporter Tag for Hidden Racemisation Journal Article
In: Synthesis (Germany), vol. 51, no. 5, pp. 1263-1272, 2019.
@article{Spránitz20191263,
title = {Organocatalytic Desymmetrisation of Fittig's Lactones: Deuterium as a Reporter Tag for Hidden Racemisation},
author = {P. Spránitz and P. Soregi and B. B. Botlik and M. Berta and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85061894028&doi=10.1055%2fs-0037-1611655&partnerID=40&md5=6145c5fd81821b592f8e8d421c65bcae},
doi = {10.1055/s-0037-1611655},
year = {2019},
date = {2019-01-01},
urldate = {2019-01-01},
journal = {Synthesis (Germany)},
volume = {51},
number = {5},
pages = {1263-1272},
abstract = {Highly enantioselective desymmetrisation of Fittig's lactones with alcohols is promoted by bifunctional cinchona squaramides. The reactions were carried out with monodeuterated methanol to detect possible hidden racemisation of the stereogenic centre. Current evidence suggests that racemisation was not a relevant process for most substrates; partial erosion of enantioselectivity was only detected with ortho -substituted aryl derivates. The resultant glutaric acid derivatives possess a scaffold that is common in natural products and the compounds are also useful chiral building blocks for further synthetic endeavours. © Georg Thieme Verlag.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

D. Fegyverneki; N. Kolozsvári; D. Molnár; O. Egyed; T. Holczbauer; T. Soós
In: Chemistry - A European Journal, vol. 25, no. 9, pp. 2100, 2019.
@article{Fegyverneki20192100,
title = {Size-Exclusion Borane-Catalyzed Domino 1,3-Allylic/Reductive Ireland–Claisen Rearrangements: Impact of the Electronic and Structural Parameters on the 1,3-Allylic Shift Aptitude},
author = {D. Fegyverneki and N. Kolozsvári and D. Molnár and O. Egyed and T. Holczbauer and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85060595026&doi=10.1002%2fchem.201900161&partnerID=40&md5=d2f67587d51747135385ea6d539326d6},
doi = {10.1002/chem.201900161},
year = {2019},
date = {2019-01-01},
urldate = {2019-01-01},
journal = {Chemistry - A European Journal},
volume = {25},
number = {9},
pages = {2100},
abstract = {Invited for the cover of this issue is the group of Tibor Soós at the Hungarian Academy of Sciences. The image depicts a metaphorical representation of the catalytic cycles and the size-exclusion concept of the reported metal-free, one-pot reductive Ireland–Claisen rearrangement. Read the full text of the article at 10.1002/chem.201805208. © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

D. Fegyverneki; N. Kolozsvári; D. Molnár; O. Egyed; T. Holczbauer; T. Soós
In: Chemistry - A European Journal, vol. 25, no. 9, pp. 2179-2183, 2019.
@article{Fegyverneki20192179,
title = {Size-Exclusion Borane-Catalyzed Domino 1,3-Allylic/Reductive Ireland–Claisen Rearrangements: Impact of the Electronic and Structural Parameters on the 1,3-Allylic Shift Aptitude},
author = {D. Fegyverneki and N. Kolozsvári and D. Molnár and O. Egyed and T. Holczbauer and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85059122430&doi=10.1002%2fchem.201805208&partnerID=40&md5=155a1237fccfb636c2b0539a4c4ebe9c},
doi = {10.1002/chem.201805208},
year = {2019},
date = {2019-01-01},
urldate = {2019-01-01},
journal = {Chemistry - A European Journal},
volume = {25},
number = {9},
pages = {2179-2183},
abstract = {The reductive Ireland–Claisen rearrangement through borane-mediated hydrosilylation is reported. The method employs a borane catalyst with a special structural design and affords access to synthetically relevant products with high diastereoselectivity. Depending on electronic and structural parameters, the reaction can be coupled with a 1,3-allylic shift, thus the valence isomer of the Ireland–Claisen product is formed. © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

2018
M. Bakos; Z. Dobi; D. Fegyverneki; Á. Gyömöre; I. Fernández; T. Soós
In: ACS Sustainable Chemistry and Engineering, vol. 6, no. 8, pp. 10869-10875, 2018.
@article{Bakos201810869,
title = {Janus Face of the Steric Effect in a Lewis Acid Catalyst with Size-Exclusion Design: Steric Repulsion and Steric Attraction in the Catalytic Exo-Selective Diels-Alder Reaction},
author = {M. Bakos and Z. Dobi and D. Fegyverneki and Á. Gyömöre and I. Fernández and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85049212179&doi=10.1021%2facssuschemeng.8b02099&partnerID=40&md5=7af36269dbf6432aeb2c089619b4c004},
doi = {10.1021/acssuschemeng.8b02099},
year = {2018},
date = {2018-01-01},
urldate = {2018-01-01},
journal = {ACS Sustainable Chemistry and Engineering},
volume = {6},
number = {8},
pages = {10869-10875},
abstract = {An exo-selective catalytic Diels-Alder reaction was developed using a Lewis acid catalyst with size-exclusion structural design. Exploiting the steric effect, especially the steric attraction, the Lewis acid catalyst was able to reroute the reaction along a higher-energy pathway. The experimental findings were also supported by theoretical calculations. This catalyst development allows an easy and practical access to highly complex and pharmaceutically relevant compounds. © 2018 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

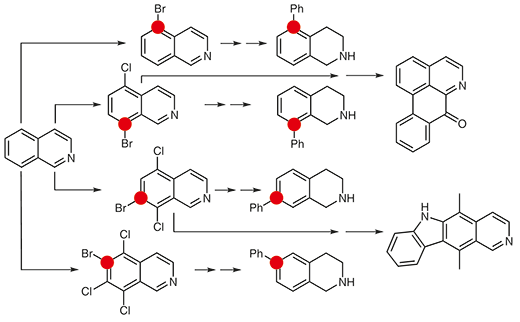
D. V. Horváth; F. Domonyi; R. Palkó; A. Lomoschitz; T. Soós
Regioexhaustive Functionalization of the Carbocyclic Core of Isoquinoline: Concise Synthesis of Oxoaporphine Core and Ellipticine Journal Article
In: Synthesis (Germany), vol. 50, no. 11, pp. 2181-2190, 2018.
@article{Horváth20182181,
title = {Regioexhaustive Functionalization of the Carbocyclic Core of Isoquinoline: Concise Synthesis of Oxoaporphine Core and Ellipticine},
author = {D. V. Horváth and F. Domonyi and R. Palkó and A. Lomoschitz and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85043311030&doi=10.1055%2fs-0037-1609153&partnerID=40&md5=1a613d40ca3ca1d2368314c74a8f5cf5},
doi = {10.1055/s-0037-1609153},
year = {2018},
date = {2018-01-01},
urldate = {2018-01-01},
journal = {Synthesis (Germany)},
volume = {50},
number = {11},
pages = {2181-2190},
abstract = {A general and versatile strategy has been developed for the functionalization of the carbocyclic core of the isoquinoline. This regioexhaustive approach employs electrophilic halogenation as a toolbox methodology and delivers highly decorated intermediates that can be further elaborated toward medicinally relevant building blocks or natural products. © Georg Thieme Verlag Stuttgart, New York.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
D. V. Horváth; T. Holczbauer; L. Bereczki; R. Palkó; N. V. May; T. Soós; P. Bombicz
Polymorphism of a porous hydrogen bond-assisted ionic organic framework Journal Article
In: CrystEngComm, vol. 20, no. 13, pp. 1779-1782, 2018.
@article{Horváth20181779,
title = {Polymorphism of a porous hydrogen bond-assisted ionic organic framework},
author = {D. V. Horváth and T. Holczbauer and L. Bereczki and R. Palkó and N. V. May and T. Soós and P. Bombicz},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85044538021&doi=10.1039%2fc8ce00041g&partnerID=40&md5=800a936524d1b62514eba32af46659d0},
doi = {10.1039/c8ce00041g},
year = {2018},
date = {2018-01-01},
urldate = {2018-01-01},
journal = {CrystEngComm},
volume = {20},
number = {13},
pages = {1779-1782},
abstract = {The polymorphism of a porous, non-covalently bonded ionic organic framework is reported. The framework is constructed by hydrogen bonding and anion⋯π interactions. In a solvatomorphic lattice, pyridine takes part in the framework formation. The role of molecular rigidity in framework construction is proven by analogous non-porous crystals, where polymorphism also appears. © 2018 The Royal Society of Chemistry.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

M. Szigeti; Z. Dobi; T. Soós
The Goldilocks Principle in Phase Labeling. Minimalist and Orthogonal Phase Tagging for Chromatography-Free Mitsunobu Reaction Journal Article
In: Journal of Organic Chemistry, vol. 83, no. 5, pp. 2869-2874, 2018.
@article{Szigeti20182869,
title = {The Goldilocks Principle in Phase Labeling. Minimalist and Orthogonal Phase Tagging for Chromatography-Free Mitsunobu Reaction},
author = {M. Szigeti and Z. Dobi and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85042881355&doi=10.1021%2facs.joc.8b00014&partnerID=40&md5=882e6c4eb9e08dcfc148dd82df552ae7},
doi = {10.1021/acs.joc.8b00014},
year = {2018},
date = {2018-01-01},
urldate = {2018-01-01},
journal = {Journal of Organic Chemistry},
volume = {83},
number = {5},
pages = {2869-2874},
abstract = {An inexpensive and chromatography-free Mitsunobu methodology has been developed using low molecular weight and orthogonally phase-tagged reagents, a tert-butyl-tagged highly apolar phosphine, and a water-soluble DIAD analogue. The byproduct of the Mitsunobu reactions can be removed by sequential liquid-liquid extractions using traditional solvents such as hexanes, MeOH, water, and EtOAc. Owing to the orthogonal phase labeling, the spent reagents can be regenerated. This new variant of the Mitsunobu reaction promises to provide an alternative and complementary solution for the well-known separation problem of the Mitsunobu reaction without having to resort to expensive, large molecular weight reagents and chromatography. © 2018 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

2017
É. Dorkó; M. Szabó; B. Kótai; I. Pápai; A. Domján; T. Soós
In: Angewandte Chemie - International Edition, vol. 56, no. 32, pp. 9512-9516, 2017.
@article{Dorkó20179512,
title = {Expanding the Boundaries of Water-Tolerant Frustrated Lewis Pair Hydrogenation: Enhanced Back Strain in the Lewis Acid Enables the Reductive Amination of Carbonyls},
author = {É. Dorkó and M. Szabó and B. Kótai and I. Pápai and A. Domján and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85021827461&doi=10.1002%2fanie.201703591&partnerID=40&md5=1cd0813ba59138a256989c559d8170fb},
doi = {10.1002/anie.201703591},
year = {2017},
date = {2017-01-01},
urldate = {2017-01-01},
journal = {Angewandte Chemie - International Edition},
volume = {56},
number = {32},
pages = {9512-9516},
abstract = {The development of a boron/nitrogen-centered frustrated Lewis pair (FLP) with remarkably high water tolerance is presented. As systematic steric tuning of the boron-based Lewis acid (LA) component revealed, the enhanced back-strain makes water binding increasingly reversible in the presence of relatively strong base. This advance allows the limits of FLP's hydrogenation to be expanded, as demonstrated by the FLP reductive amination of carbonyls. This metal-free catalytic variant displays a notably broad chemoselectivity and generality. © 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
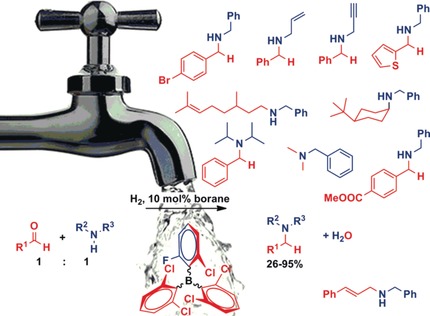
M. Bakos; Á. Gyömöre; A. Domján; T. Soós
Auto-Tandem Catalysis with Frustrated Lewis Pairs for Reductive Etherification of Aldehydes and Ketones Journal Article
In: Angewandte Chemie - International Edition, vol. 56, no. 19, pp. 5217-5221, 2017.
@article{Bakos20175217,
title = {Auto-Tandem Catalysis with Frustrated Lewis Pairs for Reductive Etherification of Aldehydes and Ketones},
author = {M. Bakos and Á. Gyömöre and A. Domján and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85017516860&doi=10.1002%2fanie.201700231&partnerID=40&md5=1b5b091e3fa74d4570d5bfcd263859c9},
doi = {10.1002/anie.201700231},
year = {2017},
date = {2017-01-01},
urldate = {2017-01-01},
journal = {Angewandte Chemie - International Edition},
volume = {56},
number = {19},
pages = {5217-5221},
abstract = {Herein we report that a single frustrated Lewis pair (FLP) catalyst can promote the reductive etherification of aldehydes and ketones. The reaction does not require an exogenous acid catalyst, but the combined action of FLP on H2, R-OH or H2O generates the required Brønsted acid in a reversible, “turn on” manner. The method is not only a complementary metal-free reductive etherification, but also a niche procedure for ethers that would be either synthetically inconvenient or even intractable to access by alternative synthetic protocols. © 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

Z. Dobi; T. Holczbauer; T. Soós
Schreiner's Thiourea Promoted [2+2] Cycloaddition of Captodative Azetidinones and Nitroolefins Journal Article
In: European Journal of Organic Chemistry, vol. 2017, no. 11, pp. 1391-1395, 2017.
@article{Dobi20171391,
title = {Schreiner's Thiourea Promoted [2+2] Cycloaddition of Captodative Azetidinones and Nitroolefins},
author = {Z. Dobi and T. Holczbauer and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85012040989&doi=10.1002%2fejoc.201601524&partnerID=40&md5=364b8d886659d577d79f3c22ead7d8e3},
doi = {10.1002/ejoc.201601524},
year = {2017},
date = {2017-01-01},
urldate = {2017-01-01},
journal = {European Journal of Organic Chemistry},
volume = {2017},
number = {11},
pages = {1391-1395},
abstract = {Strained, captodative benzylideneazetidinones were demonstrated to function as potent reaction partners in thermal [2+2] cycloadditions with nitro alkenes. The relief of strain during the cycloaddition could be leveraged to secure kinetic and thermodynamic stability for the aminonitrocyclobutane ring. Accordingly, this mild and robust procedure could be used to simplify the synthesis of azaspiro[3.3]heptanes, a motif that serves as a rigid piperidine bioisostere. © 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
![Schreiner's Thiourea Promoted [2+2] Cycloaddition of Captodative Azetidinones and Nitroolefins](http://www.orgcat.hu/wp-content/uploads/2021/10/EJOC_2017_captodative.png)
A. Bacsó; M. Szigeti; S. Varga; T. Soós
Bifunctional Thiourea-Catalyzed Stereoablative Retro-Sulfa-Michael Reaction: Concise and Diastereoselective Access to Chiral 2,4-Diarylthietanes Journal Article
In: Synthesis (Germany), vol. 49, no. 2, pp. 429-439, 2017.
@article{Bacsó2017429,
title = {Bifunctional Thiourea-Catalyzed Stereoablative Retro-Sulfa-Michael Reaction: Concise and Diastereoselective Access to Chiral 2,4-Diarylthietanes},
author = {A. Bacsó and M. Szigeti and S. Varga and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84991241342&doi=10.1055%2fs-0036-1588612&partnerID=40&md5=def2c2c9a151dcd0034822a5c7a0f8ad},
doi = {10.1055/s-0036-1588612},
year = {2017},
date = {2017-01-01},
urldate = {2017-01-01},
journal = {Synthesis (Germany)},
volume = {49},
number = {2},
pages = {429-439},
abstract = {Owing to the chiral recognition capacity of bifunctional thioureas, a stereoablative retro-sulfa-Michael reaction has been developed. Utilization of a biphasic system enabled us to render the process catalytic. The usefulness of this methodology was further illustrated by the diastereoselective synthesis of all possible stereoisomers of 2,4-diarylthiethanes. ©2017 Georg Thieme Verlag Stuttgart, New York.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

É. Dorkó; B. Kótai; T. Földes; Á. Gyömöre; I. Pápai; T. Soós
Correlating electronic and catalytic properties of frustrated Lewis pairs for imine hydrogenation Journal Article
In: Journal of Organometallic Chemistry, vol. 847, pp. 258-262, 2017.
@article{Dorkó2017258,
title = {Correlating electronic and catalytic properties of frustrated Lewis pairs for imine hydrogenation},
author = {É. Dorkó and B. Kótai and T. Földes and Á. Gyömöre and I. Pápai and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85019075536&doi=10.1016%2fj.jorganchem.2017.04.031&partnerID=40&md5=d49b6b3853d8e76131a0a6363bf2f431},
doi = {10.1016/j.jorganchem.2017.04.031},
year = {2017},
date = {2017-01-01},
urldate = {2017-01-01},
journal = {Journal of Organometallic Chemistry},
volume = {847},
pages = {258-262},
abstract = {Combined computational and experimental work to probe Lewis acidity of some boranes to be used in FLP hydrogenation. Gutmann-Beckett method of estimating Lewis acidity has limited capacity for sterically congested boranes. Calculated hydride affinity is a more appropriate tool for gauging Lewis acidity and correlate their FLP hydrogenation utility. © 2017 Elsevier B.V.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

2016
B. Berkes; K. Ozsváth; L. Molnár; T. Gáti; T. Holczbauer; G. Kardos; T. Soós
In: Chemistry - A European Journal, vol. 22, no. 50, pp. 18101-18106, 2016.
@article{Berkes201618101,
title = {Expedient and Diastereodivergent Assembly of Terpenoid Decalin Subunits having Quaternary Stereocenters through Organocatalytic Robinson Annulation of Nazarov Reagent},
author = {B. Berkes and K. Ozsváth and L. Molnár and T. Gáti and T. Holczbauer and G. Kardos and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84996549633&doi=10.1002%2fchem.201604541&partnerID=40&md5=bc5799bd8234d0a053922882fe88f0de},
doi = {10.1002/chem.201604541},
year = {2016},
date = {2016-01-01},
urldate = {2016-01-01},
journal = {Chemistry - A European Journal},
volume = {22},
number = {50},
pages = {18101-18106},
abstract = {We report an expedient approach to highly functionalized cis- and trans-decalines that could function as key structural subunits toward the synthesis of various classes of terpenoids. Key to the strategy is an organocatalyzed Robinson annulation reaction of the Nazarov reagent that affords chiral enone building blocks with high enantioselectivities. The quaternary carbon stereogenic center can direct the subsequent reactions and allow the rapid and diastereoconvergent assembly of complex decalines with contiguous stereocenters. © 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

A. Madarász; Z. Dósa; S. Varga; T. Soós; A. Csámpai; I. Pápai
Thiourea Derivatives as Brønsted Acid Organocatalysts Journal Article
In: ACS Catalysis, vol. 6, no. 7, pp. 4379-4387, 2016.
@article{Madarász20164379,
title = {Thiourea Derivatives as Brønsted Acid Organocatalysts},
author = {A. Madarász and Z. Dósa and S. Varga and T. Soós and A. Csámpai and I. Pápai},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84977090904&doi=10.1021%2facscatal.6b00618&partnerID=40&md5=28aa2b18b9c0bcc7237c5255aada5538},
doi = {10.1021/acscatal.6b00618},
year = {2016},
date = {2016-01-01},
urldate = {2016-01-01},
journal = {ACS Catalysis},
volume = {6},
number = {7},
pages = {4379-4387},
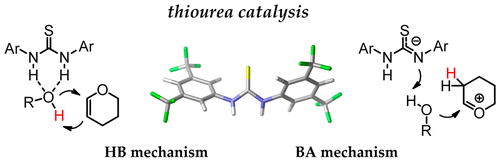
abstract = {Combined computational-experimental studies were carried out to parallel two mechanistic models for tetrahydropyranylation of alcohols catalyzed by Schreiner's thiourea. The results challenge the common mechanistic view that the catalytic effect is related to stabilizing double hydrogen-bonding interactions between the thiourea and the alcohol, which promote the attack on 3,4-dihydro-2H-pyran (DHP) (hydrogen bonding (HB) mechanism). In the alternative mechanism that we propose, thiourea acts as a Brønsted acid, protonating DHP to form an oxacarbenium ion, which reacts with the alcohol (Brønsted acid (BA) mechanism). Computations point to clear preference of transition states associated with the BA mechanism and, accordingly, predict similar catalytic activity for N-methylated thiourea and thiouracil. These predictions are confirmed experimentally. Reactions with deuterated alcohols yield both syn and anti products, providing further support for the Brønsted acid mechanism. © 2016 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
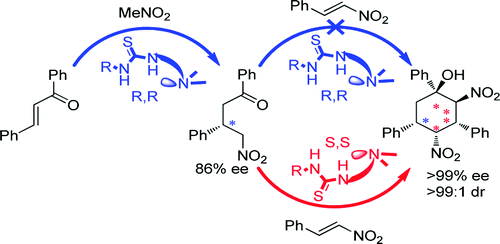
2015
S. Varga; G. Jakab; A. Csámpai; T. Soós
In: Journal of Organic Chemistry, vol. 80, no. 18, pp. 8990-8996, 2015.
@article{Varga20158990,
title = {Iterative Coupling of Two Different Enones by Nitromethane Using Bifunctional Thiourea Organocatalysts. Stereocontrolled Assembly of Cyclic and Acyclic Structures},
author = {S. Varga and G. Jakab and A. Csámpai and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84941921452&doi=10.1021%2facs.joc.5b01474&partnerID=40&md5=e414d158a193498d05ac7b40e4804287},
doi = {10.1021/acs.joc.5b01474},
year = {2015},
date = {2015-01-01},
urldate = {2015-01-01},
journal = {Journal of Organic Chemistry},
volume = {80},
number = {18},
pages = {8990-8996},
abstract = {An organocatalytic iterative assembly line has been developed in which nitromethane was sequentially coupled with two different enones using a combination of pseudoenantiomeric cinchona-based thiourea catalysts. Application of unsaturated aldehydes and ketones in the second step of the iterative sequence allows the construction of cyclic syn-ketols and acyclic compounds with multiple contiguous stereocenters. The combination of the multifunctional substrates and ambident electrophiles rendered some organocatalytic transformations possible that have not yet been realized in bifunctional noncovalent organocatalysis. © 2015 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

Á. Gyömöre; M. Bakos; T. Földes; I. Pápai; A. Domján; T. Soós
Moisture-Tolerant Frustrated Lewis Pair Catalyst for Hydrogenation of Aldehydes and Ketones Journal Article
In: ACS Catalysis, vol. 5, no. 9, pp. 5366-5372, 2015.
@article{Gyömöre20155366,
title = {Moisture-Tolerant Frustrated Lewis Pair Catalyst for Hydrogenation of Aldehydes and Ketones},
author = {Á. Gyömöre and M. Bakos and T. Földes and I. Pápai and A. Domján and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84941141930&doi=10.1021%2facscatal.5b01299&partnerID=40&md5=d447a333283463f585bb8d49a32b659c},
doi = {10.1021/acscatal.5b01299},
year = {2015},
date = {2015-01-01},
urldate = {2015-01-01},
journal = {ACS Catalysis},
volume = {5},
number = {9},
pages = {5366-5372},
abstract = {In this paper, we report on the development of a bench-stable borane for frustrated Lewis pair catalyzed reduction of aldehydes, ketones, and enones. The deliberate fine-tuning of structural and electronic parameters of Lewis acid component and the choice of Lewis base provided for the first time, a moisture-tolerant FLP catalyst. Related NMR and DFT studies underpinned the unique behavior of this FLP catalyst and gave insight into the catalytic activity of the resulting FLP catalyst. © 2015 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

Z. Dobi; T. Holczbauer; T. Soós
Strain-driven direct cross-aldol and -ketol reactions of four-membered heterocyclic ketones Journal Article
In: Organic Letters, vol. 17, no. 11, pp. 2634-2637, 2015.
@article{Dobi20152634,
title = {Strain-driven direct cross-aldol and -ketol reactions of four-membered heterocyclic ketones},
author = {Z. Dobi and T. Holczbauer and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84930628397&doi=10.1021%2facs.orglett.5b01002&partnerID=40&md5=ae5e2196949f64542e68fa673ffc0e7e},
doi = {10.1021/acs.orglett.5b01002},
year = {2015},
date = {2015-01-01},
urldate = {2015-01-01},
journal = {Organic Letters},
volume = {17},
number = {11},
pages = {2634-2637},
abstract = {Owing to the ring strain and α-heteroatom effect, the four-membered heterocyclic ketones can undergo direct cross-aldol and -ketol reactions without the need for preformed enol or "enolate-like" intermediates. Besides the organocatalyzed cross-ketol addition onto their highly active carbonyl group, their ability to act as a nucleophilic donor has also been explored. As a result, a number of discrete aldol adducts were synthesized and the distinct reactivities were successfully combined into a double-aldol one-pot reaction. © 2015 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
E. Varga; L. T. Mika; A. Csámpai; T. Holczbauer; G. Kardos; T. Soós
In: RSC Advances, vol. 5, no. 115, pp. 95079-95086, 2015.
@article{Varga201595079,
title = {Mechanistic investigations of a bifunctional squaramide organocatalyst in asymmetric Michael reaction and observation of stereoselective retro-Michael reaction},
author = {E. Varga and L. T. Mika and A. Csámpai and T. Holczbauer and G. Kardos and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84946962971&doi=10.1039%2fc5ra19593d&partnerID=40&md5=de1d64b9838202a7da151c8a0c1f6493},
doi = {10.1039/c5ra19593d},
year = {2015},
date = {2015-01-01},
urldate = {2015-01-01},
journal = {RSC Advances},
volume = {5},
number = {115},
pages = {95079-95086},
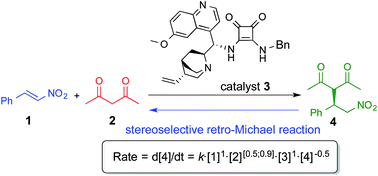
abstract = {The mechanism of the addition of acetylacetone to β-nitrostyrene catalyzed by a cinchona based squaramide catalyst was studied in detail under synthetically relevant conditions. The reaction was monitored by in situ IR and 1H-NMR spectroscopy and a reaction mechanism was proposed based on these kinetics experiments. It was found that the reaction shows nearly first order dependence on both substrates and catalyst. Our investigations also revealed that the catalyst was able to promote stereoselective retro-Michael reaction. © 2015 The Royal Society of Chemistry.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
A. Jakab; Z. Dalicsek; T. Holczbauer; A. Hamza; I. Pápai; Z. Finta; G. Timári; T. Soós
Superstable palladium(0) complex as an air-and thermostable catalyst for Suzuki coupling reactions Journal Article
In: European Journal of Organic Chemistry, vol. 2015, no. 1, pp. 60-66, 2015.
@article{Jakab201560,
title = {Superstable palladium(0) complex as an air-and thermostable catalyst for Suzuki coupling reactions},
author = {A. Jakab and Z. Dalicsek and T. Holczbauer and A. Hamza and I. Pápai and Z. Finta and G. Timári and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84919678100&doi=10.1002%2fejoc.201403214&partnerID=40&md5=29da73f7df96b15d367af688b80899b4},
doi = {10.1002/ejoc.201403214},
year = {2015},
date = {2015-01-01},
urldate = {2015-01-01},
journal = {European Journal of Organic Chemistry},
volume = {2015},
number = {1},
pages = {60-66},
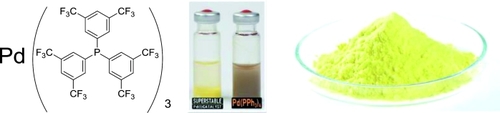
abstract = {An unprecedentedly thermo-and air-stable Pd0 complex from readily available electron-poor trifluoromethylated phosphine was serendipitously discovered. As detailed and comparative DFT calculations indicate, the stability of the complex is associated with unusually strong ligand-ligand noncovalent interactions. The unique stability and the presence of hydrophobic structural elements of the complex offer several practical advantages, which were exploited in catalytic Suzuki-Miyaura coupling reactions. © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
A. Jakab; Z. Dalicsek; T. Soós
A robust and efficient catalyst possessing an electron-deficient ligand for the palladium-catalyzed direct arylation of heteroarenes Journal Article
In: European Journal of Organic Chemistry, vol. 2015, no. 1, pp. 56-59, 2015.
@article{Jakab201556,
title = {A robust and efficient catalyst possessing an electron-deficient ligand for the palladium-catalyzed direct arylation of heteroarenes},
author = {A. Jakab and Z. Dalicsek and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84919346818&doi=10.1002%2fejoc.201402586&partnerID=40&md5=21e77b27faf59a1d4037a36a402fb359},
doi = {10.1002/ejoc.201402586},
year = {2015},
date = {2015-01-01},
urldate = {2015-01-01},
journal = {European Journal of Organic Chemistry},
volume = {2015},
number = {1},
pages = {56-59},
abstract = {The exploration of the direct arylation capacity of a unique, thermally stable, and air-stable Pd0-phosphine catalyst is reported. Besides decisively contributing to catalyst robustness, the electron-deficient trifluoromethyl-substituted triphenylphosphine ligands make the palladium center more electron-deficient and accelerate the direct arylation step. The combination of only 0.5-2 mol-% of the catalyst and a substoichiometric quantity of pivalic acid generates an efficient system to promote biaryl-forming reactions of a broad range of electronically varied hetarenes and aryl bromides. The observed regioselective arylations suggest that a concerted metalation-deprotonation pathway is involved in the C-H activation step. © 2014 SIOC, CAS, Shanghai, & WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

2014
E. Gráczer; A. Bacsó; D. Kónya; A. Kazi; T. Soós; L. Molnár; T. Szimler; L. Beinrohr; A. Szilágyi; P. Závodszky; M. Vas
Drugs against Mycobacterium tuberculosis 3-isopropylmalate dehydrogenase can be developed using homologous enzymes as surrogate targets Journal Article
In: Protein and Peptide Letters, vol. 21, no. 12, pp. 1295-1307, 2014.
@article{Gráczer20141295,
title = {Drugs against Mycobacterium tuberculosis 3-isopropylmalate dehydrogenase can be developed using homologous enzymes as surrogate targets},
author = {E. Gráczer and A. Bacsó and D. Kónya and A. Kazi and T. Soós and L. Molnár and T. Szimler and L. Beinrohr and A. Szilágyi and P. Závodszky and M. Vas},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84925642604&partnerID=40&md5=12f4e1ab54b114e2f4502ac5af2782dd},
year = {2014},
date = {2014-01-01},
journal = {Protein and Peptide Letters},
volume = {21},
number = {12},
pages = {1295-1307},
abstract = {3-Isopropylmalate dehydrogenase (IPMDH) from Mycobacterium tuberculosis (Mtb) may be a target for specific drugs against this pathogenic bacterium. We have expressed and purified Mtb IPMDH and determined its physicalchemical and enzymological properties. Size-exclusion chromatography and dynamic light scattering measurements (DLS) suggest a tetrameric structure for Mtb IPMDH, in contrast to the dimeric structure of most IPMDHs. The kinetic properties (kcat and Km values) of Mtb IPMDH and the pH-dependence of kcat are very similar to both Escherichia coli (Ec) and Thermus thermophilus (Tt) IPMDHs. The stability of Mtb IPMDH in 8 M urea is close to that of the mesophilic counterpart, Ec IPMDH, both of them being much less stable than the thermophilic (Tt) enzyme. Two known IPMDH inhibitors, O-methyl oxalohydroxamate and 3-methylmercaptomalate, have been synthesised. Their inhibitory effects were found to be independent of the origin of IPMDHs. Thus, experiments with either Ec or Tt IPMDH would be equally relevant for designing specific inhibitory drugs against Mtb IPMDH. © 2014 Bentham Science Publishers.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
G. M. Keserű; T. Soós; C. O. Kappe
Anthropogenic reaction parameters-the missing link between chemical intuition and the available chemical space Journal Article
In: Chemical Society Reviews, vol. 43, no. 15, pp. 5387-5399, 2014.
@article{Keser20145387,
title = {Anthropogenic reaction parameters-the missing link between chemical intuition and the available chemical space},
author = {G. M. Keserű and T. Soós and C. O. Kappe},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84903954633&doi=10.1039%2fc3cs60423c&partnerID=40&md5=88fd52134ba91431e5707402e2f8a526},
doi = {10.1039/c3cs60423c},
year = {2014},
date = {2014-01-01},
urldate = {2014-01-01},
journal = {Chemical Society Reviews},
volume = {43},
number = {15},
pages = {5387-5399},
abstract = {How do skilled synthetic chemists develop good intuitive expertise? Why can we only access such a small amount of the available chemical space - both in terms of the reactions used and the chemical scaffolds we make? We argue here that these seemingly unrelated questions have a common root and are strongly interdependent. We performed a comprehensive analysis of organic reaction parameters dating back to 1771 and discovered that there are several anthropogenic factors that limit reaction parameters and thus the scope of synthetic chemistry. Nevertheless, many of the anthropogenic limitations such as narrow parameter space and the opportunity for rapid and clear feedback on the progress of reactions appear to be crucial for the acquisition of valid and reliable chemical intuition. In parallel, however, all of these same factors represent limitations for the exploration of available chemistry space and we argue that these are thus at least partly responsible for limited access to new chemistries. We advocate, therefore, that the present anthropogenic boundaries can be expanded by a more conscious exploration of "off-road" chemistry that would also extend the intuitive knowledge of trained chemists. This journal is © the Partner Organisations 2014.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

B. Kótai; G. Kardos; A. Hamza; V. Farkas; I. Pápai; T. Soós
On the mechanism of bifunctional squaramide-catalyzed organocatalytic michael addition: A protonated catalyst as an oxyanion hole Journal Article
In: Chemistry - A European Journal, vol. 20, no. 19, pp. 5631-5639, 2014.
@article{Kótai20145631,
title = {On the mechanism of bifunctional squaramide-catalyzed organocatalytic michael addition: A protonated catalyst as an oxyanion hole},
author = {B. Kótai and G. Kardos and A. Hamza and V. Farkas and I. Pápai and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84899933905&doi=10.1002%2fchem.201304553&partnerID=40&md5=c0b344aa41b8af0e6f89c17e175b03c6},
doi = {10.1002/chem.201304553},
year = {2014},
date = {2014-01-01},
urldate = {2014-01-01},
journal = {Chemistry - A European Journal},
volume = {20},
number = {19},
pages = {5631-5639},
abstract = {A joint experimental-theoretical study of a bifunctional squaramide-amine-catalyzed Michael addition reaction between 1,3-dioxo nucleophiles and nitrostyrene has been undertaken to gain insight into the nature of bifunctional organocatalytic activation. For this highly stereoselective reaction, three previously proposed mechanistic scenarios for the critical C-C bond-formation step were examined. Accordingly, the formation of the major stereoisomeric products is most plausible by one of the bifunctional pathways that involve electrophile activation by the protonated amine group of the catalyst. However, some of the minor product isomers are also accessible through alternative reaction routes. Structural analysis of transition states points to the structural invariance of certain fragments of the transition state, such as the protonated catalyst and the anionic fragment of approaching reactants. Our topological analysis provides deeper insight and a more general understanding of bifunctional noncovalent organocatalysis. Finding the path: The mechanism of bifunctional squaramide-promoted Michael addition of prochiral 1,3-dioxo nucleophiles and nitroolefin has been studied on the basis of DFT calculations. Among the investigated mechanistic scenarios, the pathway corresponding to electrophile activation via the protonated amine unit is found to be the most feasible (see figure). For some of the minor stereoisomeric products, alternative pathways are also accessible. © 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

É. Dorkó; E. Varga; T. Gáti; T. Holczbauer; I. Pápai; H. Mehdi; T. Soós
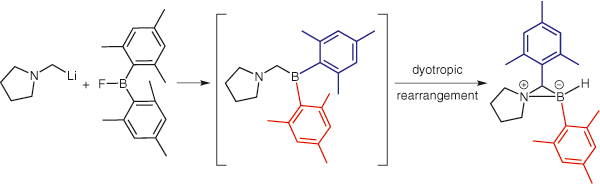
Steric control of geminal Lewis pair behavior: Frustration induced dyotropic rearrangement Journal Article
In: Synlett, vol. 25, no. 11, pp. 1525-1528, 2014.
@article{Dorkó20141525,
title = {Steric control of geminal Lewis pair behavior: Frustration induced dyotropic rearrangement},
author = {É. Dorkó and E. Varga and T. Gáti and T. Holczbauer and I. Pápai and H. Mehdi and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84903269245&doi=10.1055%2fs-0033-1339125&partnerID=40&md5=7c0e8209e625faf2a39580633d6ed9dd},
doi = {10.1055/s-0033-1339125},
year = {2014},
date = {2014-01-01},
urldate = {2014-01-01},
journal = {Synlett},
volume = {25},
number = {11},
pages = {1525-1528},
abstract = {A series of methylene-linked boron/nitrogen geminal Lewis pairs were synthesized and the impacts of sterical effect on their chemical behavior were systematically investigated. Increasing the steric demand around the boron atom is manifested first by an incremental change in the structure of the resulting dative adducts. Accordingly, in the case of phenyl substituents (Alk 2NCH2BPh2), formation of head-to-tail dimers/oligomers was observed, while such an intermolecular association was avoided when o-tolyl moiety was introduced [Alk2NCH 2B(o-Tol)2], affording only an intramolecular dative complex. Further increase of sterical hindrance to a point (i.e. using mesityl substituents), however, caused a radical change in the structure; a dyotropic rearrangement occurred. Thus, steric interference induced a rearrangement in the geminal pair to relieve or minimize the frustration strain. © Georg Thieme Verlag Stuttgart New York.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2013
A. Demeter; K. Horváth; K. Böor; L. Molnár; T. Soós; G. Lendvay
Substituent effect on the photoreduction kinetics of benzophenone Journal Article
In: Journal of Physical Chemistry A, vol. 117, no. 40, pp. 10196-10210, 2013.
@article{Demeter201310196,
title = {Substituent effect on the photoreduction kinetics of benzophenone},
author = {A. Demeter and K. Horváth and K. Böor and L. Molnár and T. Soós and G. Lendvay},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84885648841&doi=10.1021%2fjp406269e&partnerID=40&md5=d1851c7b010b7e3dfb78a3417246ba12},
doi = {10.1021/jp406269e},
year = {2013},
date = {2013-01-01},
urldate = {2013-01-01},
journal = {Journal of Physical Chemistry A},
volume = {117},
number = {40},
pages = {10196-10210},
abstract = {The kinetics of the photoreduction of four benzophenone derivatives by isopropyl alcohol was examined in acetonitrile, namely, tetra-meta- trifluoromethyl-, di-para-trifluoromethyl-, di-para-methoxy benzophenone, and, for comparison, the unsubstituted molecule itself. The basic spectroscopic (absorption and phosphorescence spectra) and photophysical (quantum yields and excited state energies) properties were established, and the key kinetic parameters were determined by the laser flash photolysis transient absorption technique. The rate coefficients of both the primary and secondary photoreduction reaction show remarkable dependence on ring substitution. This substantial effect is caused by the considerable change in the activation energy of the corresponding process. The experimental results as well as DFT quantum chemical calculations clearly indicate that these benzophenone derivatives all react as n-π* excited ketones, and the rate as well as the activation energy of the reduction steps change parallel with the reaction enthalpies, the determining factor being the stability of the forming aromatic ketyl radicals. The secondary photoreduction of benzophenones by the aliphatic ketyl radical formed in the primary step occurs via a hydrogen bonded complex. The binding energy of the hydrogen bonded complex between the aliphatic ketyl radical reactant and a solvent molecule is a critical parameter influencing the observable rate of the secondary photoreduction. © 2013 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

G. Kardos; T. Soós
Tether-free immobilized bifunctional squaramide organocatalysts for batch and flow reactions Journal Article
In: European Journal of Organic Chemistry, no. 21, pp. 4490-4494, 2013.
@article{Kardos20134490,
title = {Tether-free immobilized bifunctional squaramide organocatalysts for batch and flow reactions},
author = {G. Kardos and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84880044955&doi=10.1002%2fejoc.201300626&partnerID=40&md5=b19ef11a09733aaead038d82cc254e58},
doi = {10.1002/ejoc.201300626},
year = {2013},
date = {2013-01-01},
urldate = {2013-01-01},
journal = {European Journal of Organic Chemistry},
number = {21},
pages = {4490-4494},
abstract = {This paper describes the preparation of highly efficient, easily accessible, and robust immobilized bifunctional organocatalysts. There was no need to employ any tether to secure high enantio- and diastereoselectivities in various Michael addition reactions. The synthetically useful Michael adducts were obtained within reasonable reaction times with the advantage of easy product isolation and the possibility of automation by using a flow chemistry apparatus. Easily accessible, robust, and cheap immobilized organocatalysts are developed and used to prepare Michael adducts in excellent yields with excellent enantioselectivities, even on the gram scale. Copyright © 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

2012
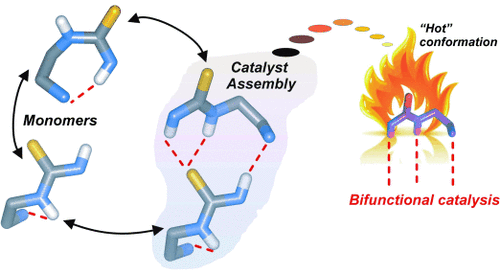
G. Tárkányi; P. Király; T. Soós; S. Varga
Active conformation in amine-thiourea bifunctional organocatalysis preformed by catalyst aggregation Journal Article
In: Chemistry - A European Journal, vol. 18, no. 7, pp. 1918-1922, 2012.
@article{Tárkányi20121918,
title = {Active conformation in amine-thiourea bifunctional organocatalysis preformed by catalyst aggregation},
author = {G. Tárkányi and P. Király and T. Soós and S. Varga},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84856695332&doi=10.1002%2fchem.201102701&partnerID=40&md5=338aa8ee97287b2b50d03be84ec97856},
doi = {10.1002/chem.201102701},
year = {2012},
date = {2012-01-01},
urldate = {2012-01-01},
journal = {Chemistry - A European Journal},
volume = {18},
number = {7},
pages = {1918-1922},
abstract = {Self-activation: Takemoto's catalyst gains access to its active conformation by equilibrating between its hydrogen-bonded intra- and intermolecular interactions in apolar aprotic solvents. By destabilization of the inactive monomeric conformations, the extended anti-anti thiourea conformation is preformed in the assembly. On leaving the assembly, this transient conformation has a structural preference to become a catalytically active monomeric species that has the potency for dual activation (see scheme). © 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
G. Erős; K. Nagy; H. Mehdi; I. Pápai; P. Nagy; P. Király; G. Tárkányi; T. Soós
Catalytic hydrogenation with frustrated lewis pairs: Selectivity achieved by size-exclusion design of lewis acids Journal Article
In: Chemistry - A European Journal, vol. 18, no. 2, pp. 574-585, 2012.
@article{Erös2012574,
title = {Catalytic hydrogenation with frustrated lewis pairs: Selectivity achieved by size-exclusion design of lewis acids},
author = {G. Erős and K. Nagy and H. Mehdi and I. Pápai and P. Nagy and P. Király and G. Tárkányi and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84855327166&doi=10.1002%2fchem.201102438&partnerID=40&md5=0711c36e47345a8434c9258412f6c1a5},
doi = {10.1002/chem.201102438},
year = {2012},
date = {2012-01-01},
urldate = {2012-01-01},
journal = {Chemistry - A European Journal},
volume = {18},
number = {2},
pages = {574-585},
abstract = {Catalytic hydrogenation that utilizes frustrated Lewis pair (FLP) catalysts is a subject of growing interest because such catalysts offer a unique opportunity for the development of transition-metal-free hydrogenations. The aim of our recent efforts is to further increase the functional-group tolerance and chemoselectivity of FLP catalysts by means of size-exclusion catalyst design. Given that hydrogen molecule is the smallest molecule, our modified Lewis acids feature a highly shielded boron center that still allows the cleavage of the hydrogen but avoids undesirable FLP reactivity by simple physical constraint. As a result, greater latitude in substrate scope can be achieved, as exemplified by the chemoselective reduction of α,β-unsaturated imines, ketones, and quinolines. In addition to synthetic aspects, detailed NMR spectroscopic, DFT, and 2H isotopic labeling studies were performed to gain further mechanistic insight into FLP hydrogenation. Copyright © 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

2011
T. Soós
Design of frustrated lewis pair catalysts for metal-free and selective hydrogenation Journal Article
In: Pure and Applied Chemistry, vol. 83, no. 3, pp. 667-675, 2011.
@article{Sós2011667,
title = {Design of frustrated lewis pair catalysts for metal-free and selective hydrogenation},
author = {T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-79951833533&doi=10.1351%2fPAC-CON-11-01-02&partnerID=40&md5=794fb72949cd5596ebf9f8d07f9f4087},
doi = {10.1351/PAC-CON-11-01-02},
year = {2011},
date = {2011-01-01},
urldate = {2011-01-01},
journal = {Pure and Applied Chemistry},
volume = {83},
number = {3},
pages = {667-675},
abstract = {After a short introduction to place our work in a proper context, this account summarizes our theoretical and synthetic results in the field of frustrated Lewis pair (FLP) chemistry. © 2011 IUPAC.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
S. Varga; G. Jakab; L. Drahos; T. Holczbauer; M. Czugler; T. Soós
In: Organic Letters, vol. 13, no. 20, pp. 5416-5419, 2011.
@article{Varga20115416,
title = {Double diastereocontrol in bifunctional thiourea organocatalysis: Iterative Michael-Michael-Henry sequence regulated by the configuration of chiral catalysts},
author = {S. Varga and G. Jakab and L. Drahos and T. Holczbauer and M. Czugler and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-80054695939&doi=10.1021%2fol201559j&partnerID=40&md5=8884f1f7610d8c16e18a8a3ab873a1a0},
doi = {10.1021/ol201559j},
year = {2011},
date = {2011-01-01},
urldate = {2011-01-01},
journal = {Organic Letters},
volume = {13},
number = {20},
pages = {5416-5419},
abstract = {The importance and reactivity consequences of the double diastereocontrol in noncovalent bifunctional organocatalysis were studied. The results suggest that the bifunctional thioureas can have synthetic limitations in multicomponent domino or autotandem catalysis. Nevertheless, we provided a means to exploit this behavior and used the configuration of the chiral catalyst as a control element in organo-sequential reactions. © 2011 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2010
G. Erős; H. Mehdi; I. Pápai; T. A. Rokob; P. Király; G. Tárkányi; T. Soós
Expanding the scope of metal-free catalytic hydrogenation through frustrated Lewis pair design Journal Article
In: Angewandte Chemie - International Edition, vol. 49, no. 37, pp. 6559-6563, 2010.
@article{Eros20106559,
title = {Expanding the scope of metal-free catalytic hydrogenation through frustrated Lewis pair design},
author = {G. Erős and H. Mehdi and I. Pápai and T. A. Rokob and P. Király and G. Tárkányi and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-77956525329&doi=10.1002%2fanie.201001518&partnerID=40&md5=1f3aff65566f6f7377cb822a1c6aa63c},
doi = {10.1002/anie.201001518},
year = {2010},
date = {2010-01-01},
urldate = {2010-01-01},
journal = {Angewandte Chemie - International Edition},
volume = {49},
number = {37},
pages = {6559-6563},
abstract = {No metal causes frustration: Based on a conceptual framework, novel frustrated Lewis acid-base catalyst systems with orthogonal reactivity have been developed. Aside from enhanced functional-group tolerance, unique chemoselectivity can be achieved in catalytic metal-free hydrogenations (see scheme). © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

P. Király; T. Soós; S. Varga; B. Vakulya; G. Tárkányi
Self-association promoted conformational transition of (3R,4S,8R,9R)-9-[(3, 5-bis(trifluoromethyl)phenyl))-thiourea](9-deoxy)-epi-cinchonine Journal Article
In: Magnetic Resonance in Chemistry, vol. 48, no. 1, pp. 13-19, 2010.
@article{Király201013,
title = {Self-association promoted conformational transition of (3R,4S,8R,9R)-9-[(3, 5-bis(trifluoromethyl)phenyl))-thiourea](9-deoxy)-epi-cinchonine},
author = {P. Király and T. Soós and S. Varga and B. Vakulya and G. Tárkányi},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-73949129298&doi=10.1002%2fmrc.2531&partnerID=40&md5=ef682695ad1ce72ce06f871a043a3b4b},
doi = {10.1002/mrc.2531},
year = {2010},
date = {2010-01-01},
urldate = {2010-01-01},
journal = {Magnetic Resonance in Chemistry},
volume = {48},
number = {1},
pages = {13-19},
abstract = {The conformational diversity of the (3R,4S,8R,9R)-9-[(3,5- bis(trifluoromethyl)phenyl))-thiourea](9-deoxy)-epi-cinchonine organocatalyst is discussed. Low-temperature NMR experiments confirmed a self-association process, which promotes the quinoline rotation between two intramolecularly hydrogen-bonded monomeric conformers of the catalyst. The balanced population of the coexisting monomeric and dimeric species allowed us to conduct a structural study of a rather complex conformational dynamics of the pure catalyst. The study is extended by a comparison with other members of the bifunctional amine-thiourea organocatalyst family. Changes in themolecular structure of the catalysts influence the interplaybetween intraand intermolecular hydrogen bonding, and yield different extent of catalyst self-association. By assessing the conformation of the individual states, we established the thermodynamic model of a self-association promoted conformational transition. Copyright © 2009 John Wiley & Sons, Ltd.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
-epi-cinchonine](http://www.orgcat.hu/wp-content/uploads/2021/10/MRC_2010_dimer.jpg)
2009
T. Soós
Fluorous Chiral Catalyst Immobilization Book
2009.
Links | Tags:
@book{Soós2009179,
title = {Fluorous Chiral Catalyst Immobilization},
author = {T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84889438157&doi=10.1002%2f9780470682005.ch8&partnerID=40&md5=665e1226b960064d54084b0310d18260},
doi = {10.1002/9780470682005.ch8},
year = {2009},
date = {2009-01-01},
journal = {Recoverable and Recyclable Catalysts},
pages = {179-198},
keywords = {},
pubstate = {published},
tppubtype = {book}
}
Z. Dalicsek; F. Pollreisz; T. Soós
Efficient separation of a trifluoromethyl substituted organocatalyst: Just add water Journal Article
In: Chemical Communications, no. 30, pp. 4587-4589, 2009.
@article{Dalicsek20094587,
title = {Efficient separation of a trifluoromethyl substituted organocatalyst: Just add water},
author = {Z. Dalicsek and F. Pollreisz and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-68149141430&doi=10.1039%2fb908967e&partnerID=40&md5=1fa47dadf9c80158de874bdc75d0de75},
doi = {10.1039/b908967e},
year = {2009},
date = {2009-01-01},
urldate = {2009-01-01},
journal = {Chemical Communications},
number = {30},
pages = {4587-4589},
abstract = {A practical, cost-saving tagging approach is developed which takes advantage of the hydrophobicity of trifluoromethyl groups, exemplified by the application and recovery of a CBS precatalyst using tuned aqueous-organic media with minimum 50% water content. © The Royal Society of Chemistry 2009.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

2008
G. Tárkányi; P. Király; S. Varga; B. Vakulya; T. Soós
Edge-to-face CH/π aromatic interaction and molecular self-recognition in epi-cinchona-based bifunctional thiourea organocatalysis Journal Article
In: Chemistry - A European Journal, vol. 14, no. 20, pp. 6078-6086, 2008.
@article{Tárkányi20086078,
title = {Edge-to-face CH/π aromatic interaction and molecular self-recognition in epi-cinchona-based bifunctional thiourea organocatalysis},
author = {G. Tárkányi and P. Király and S. Varga and B. Vakulya and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-53849086148&doi=10.1002%2fchem.200800197&partnerID=40&md5=793cd2313eaaada5b1ee338807489713},
doi = {10.1002/chem.200800197},
year = {2008},
date = {2008-01-01},
urldate = {2008-01-01},
journal = {Chemistry - A European Journal},
volume = {14},
number = {20},
pages = {6078-6086},
abstract = {The impact of cooperativity between intermolecular interactions is demonstrated by the molecular self-recognition properties of highly enantioselective epi-cinchona bifunctional thiourea organocatalysts. Low-temperature NMR experiments in inert solvents have revealed two sets of nonequivalent resonances in equal population for thiourea-modified members of the epi-quinine and epi-quinidine families. In solution, the predominance of an asymmetric (C1) dimeric self-assembly with noteworthy structural motifs became evident: simultaneous intra-and intermolecular thiourea hydrogen bonding and a CH/π interaction were observed. Both the stereochemical and the diverse conformational features of the system favor the observed quinoline T-shaped aromatic π-π stacking interaction. The structure findings are supported by quantitative protonproton distance data that were available from NOE buildup curves. The 3D structure of the dimeric assembly has been modeled in agreement with the H-H distance restraints. Owing to the geometrical preference associated with the dimerization process, the self-assembled bifunctional system is interpreted as a charge-transfer complex with the potential for catalyst self-activation. © 2008 Wiley-VCH Verlag GmbH & Co. KGaA.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

B. Vakulya; S. Varga; T. Soós
In: Journal of Organic Chemistry, vol. 73, no. 9, pp. 3475-3480, 2008.
@article{Vakulya20083475,
title = {Epi-cinchona based thiourea organocatalyst family as an efficient asymmetric Michael addition promoter: Enantioselective conjugate addition of nitroalkanes to chalcones and α,β-unsaturated N-acylpyrroles},
author = {B. Vakulya and S. Varga and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-43449135616&doi=10.1021%2fjo702692a&partnerID=40&md5=84615d7c24ff15f0956458d5994bf5be},
doi = {10.1021/jo702692a},
year = {2008},
date = {2008-01-01},
urldate = {2008-01-01},
journal = {Journal of Organic Chemistry},
volume = {73},
number = {9},
pages = {3475-3480},
abstract = {(Chemical Equation Presented) A small set of easily available epi-cinchona based thiourea organocatalysts have been synthesized and tested in enantioselective Michael addition of nitroalkanes to chalcones. These bifunctional catalyst systems promoted the conjugate additions with high enantioselectivities and chemical yields. The extension of this methodology was further explored to encompass α,β-unsaturated N-acylpyrroles, as a chalcone mimic. Functionally, the N-acylpyrrole moiety in the adduct acts as an ester surrogate; therefore, it can easily be transformed to various valuable and biologically relevant compounds. This approach allowed the concise stereoselective synthesis of (R)-rolipram. © 2008 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

T. A. Rokob; A. Hamza; A. Stirling; T. Soós; I. Pápai
Turning frustration into bond activation: A theoretical mechanistic study on heterolytic hydrogen splitting by frustrated Lewis pairs Journal Article
In: Angewandte Chemie - International Edition, vol. 47, no. 13, pp. 2435-2438, 2008.
@article{Rokob20082435,
title = {Turning frustration into bond activation: A theoretical mechanistic study on heterolytic hydrogen splitting by frustrated Lewis pairs},
author = {T. A. Rokob and A. Hamza and A. Stirling and T. Soós and I. Pápai},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-42449158036&doi=10.1002%2fanie.200705586&partnerID=40&md5=8bf4ce19ec2b5b48260bd065b3493160},
doi = {10.1002/anie.200705586},
year = {2008},
date = {2008-01-01},
urldate = {2008-01-01},
journal = {Angewandte Chemie - International Edition},
volume = {47},
number = {13},
pages = {2435-2438},
abstract = {(Figure Presented) Just before splitting: A mechanistic model has been proposed for H2 activation by sterically demanding phosphine-borane Lewis pairs. There is theoretical evidence for noncovalent intermolecular association of donor-acceptor molecules to form a flexible but energetically strained complex, which provides preorganized active centers for heterolytic H-H bond cleavage (see picture). © 2008 Wiley-VCH Verlag GmbH & Co. KGaA.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

2006
A. Hamza; G. Schubert; T. Soós; I. Pápai
Theoretical studies on the bifunctionality of chiral thiourea-based organocatalysts: Competing routes to C-C bond formation Journal Article
In: Journal of the American Chemical Society, vol. 128, no. 40, pp. 13151-13160, 2006.
@article{Hamza200613151,
title = {Theoretical studies on the bifunctionality of chiral thiourea-based organocatalysts: Competing routes to C-C bond formation},
author = {A. Hamza and G. Schubert and T. Soós and I. Pápai},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84962467066&doi=10.1021%2fja063201x&partnerID=40&md5=8411c051899d73d935800760e6ae5e9f},
doi = {10.1021/ja063201x},
year = {2006},
date = {2006-01-01},
urldate = {2006-01-01},
journal = {Journal of the American Chemical Society},
volume = {128},
number = {40},
pages = {13151-13160},
abstract = {The mechanism of enantioselective Michael addition of acetylacetone to a nitroolefin catalyzed by a thiourea-based chiral bifunctional organocatalyst is investigated using density functional theory calculations. A systematic conformational analysis is presented for the catalyst, and it is shown that both substrates coordinate preferentially via bidentate hydrogen bonds. The deprotonation of the enol form of acetylacetone by the amine of the catalyst is found to occur easily, leading to an ion pair characterized by multiple H-bonds involving the thiourea unit as well. Two distinct reaction pathways are explored toward the formation of the Michael product that differ in the mode of electrophile activation. Both reaction channels are shown to be consistent with the notion of noncovalent organocatalysis in that the transition states leading to the Michael adduct are stabilized by extensive H-bonded networks. The comparison of the obtained energetics for the two pathways allows us to propose an alternative mechanistic rationale for asymmetric C-C bond forming reactions catalyzed by bifunctional thiourea derivatives. The origin of enantioselectivity in the investigated reaction is also discussed. © 2006 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

Z. Kaleta; B. T. Makowski; T. Soós; R. Dembinski
Thionation using fluorous Lawesson's reagent Journal Article
In: Organic Letters, vol. 8, no. 8, pp. 1625-1628, 2006.
@article{Kaleta20061625,
title = {Thionation using fluorous Lawesson's reagent},
author = {Z. Kaleta and B. T. Makowski and T. Soós and R. Dembinski},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-33646458226&doi=10.1021%2fol060208a&partnerID=40&md5=4fa7d1d90fa054b205e6317ef4fc7ef4},
doi = {10.1021/ol060208a},
year = {2006},
date = {2006-01-01},
urldate = {2006-01-01},
journal = {Organic Letters},
volume = {8},
number = {8},
pages = {1625-1628},
abstract = {Thionation of amides, 1,4-diketones, N-(2-oxoalkyl)amides, N,N-acylhydrazines, and acyl-protected uridines with the use of a fluorous analogue of the Lawesson's reagent leads to thioamides, thiophenes, 1,3-thiazoles, 1,3,4-thiadiazoles, and acyl-protected 4-thiouridines. The isolation of the final products in high yields is achieved in most cases by a simple filtration (fluorous solid-phase extraction). © 2006 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

Z. Kaleta; G. Tárkányi; Á. Gömöry; F. Kálmán; T. Nagy; T. Soós
Synthesis and application of a fluorous Lawesson's reagent: Convenient chromatography-free product purification Journal Article
In: Organic Letters, vol. 8, no. 6, pp. 1093-1095, 2006.
@article{Kaleta20061093,
title = {Synthesis and application of a fluorous Lawesson's reagent: Convenient chromatography-free product purification},
author = {Z. Kaleta and G. Tárkányi and Á. Gömöry and F. Kálmán and T. Nagy and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-33646441438&doi=10.1021%2fol0529849&partnerID=40&md5=bb77ab1b971db9de529ed7127d66e9e4},
doi = {10.1021/ol0529849},
year = {2006},
date = {2006-01-01},
urldate = {2006-01-01},
journal = {Organic Letters},
volume = {8},
number = {6},
pages = {1093-1095},
abstract = {A fluorous analogue of Lawesson's reagent for thionation of carbonyl compounds has been developed and its use demonstrated on a series of amides, esters, and ketones. The separation of the Lawesson's reagent-derived byproducts can be achieved by a simple fluorous solid-phase extraction. © 2006 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

2005
Z. Dalicsek; F. Pollreisz; Á. Gömöry; T. Soós
Recoverable fluorous CBS methodology for asymmetric reduction of ketones Journal Article
In: Organic Letters, vol. 7, no. 15, pp. 3243-3246, 2005.
@article{Dalicsek20053243,
title = {Recoverable fluorous CBS methodology for asymmetric reduction of ketones},
author = {Z. Dalicsek and F. Pollreisz and Á. Gömöry and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-23044509130&doi=10.1021%2fol051024j&partnerID=40&md5=8455a11a3103d76c24c729eccb2a0c54},
doi = {10.1021/ol051024j},
year = {2005},
date = {2005-01-01},
urldate = {2005-01-01},
journal = {Organic Letters},
volume = {7},
number = {15},
pages = {3243-3246},
abstract = {(Chemical Equation Presented) An operationally simple and recoverable fluorous CBS methodology was developed. The in situ-generated fluorous oxazaborolidine efficiently catalyzed the reduction of ketones with high enantioselectivity and reactivity. The subsequent recycling of the fluorous prolinol precatalyst was achieved by fluorous solid-phase extraction. © 2005 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

Z. Kaleta; O. Egyed; T. Soós
Fluorous click chemistry as a practical tagging method Journal Article
In: Organic and Biomolecular Chemistry, vol. 3, no. 12, pp. 2228-2230, 2005.
@article{Kaleta20052228,
title = {Fluorous click chemistry as a practical tagging method},
author = {Z. Kaleta and O. Egyed and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-23344445780&doi=10.1039%2fb504973c&partnerID=40&md5=f9effbc19ae5b53e6db0cccb0bab393c},
doi = {10.1039/b504973c},
year = {2005},
date = {2005-01-01},
urldate = {2005-01-01},
journal = {Organic and Biomolecular Chemistry},
volume = {3},
number = {12},
pages = {2228-2230},
abstract = {A fluorous version of azide click chemistry was developed and its utility was demonstrated in the synthesis of the fluorous cinchona alkaloid. Highly efficient fluorous tagging methodology was developed based on catalytic 1,3-dipolar cycloaddition as the key step. This method resulted in high yield, utilized mild reaction conditions and had a high functional group tolerance and chemoselectivity. The successful application of click chemistry to fluorous chemistry gave an impetus to demonstrate its utility in the fluorous synthesis of a more complex molecule. The fluorous tagging procedure offered an improvement in both efficiency and practical simplicity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

B. Vakulya; S. Varga; A. Csámpai; T. Soós
Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts Journal Article
In: Organic Letters, vol. 7, no. 10, pp. 1967-1969, 2005.
@article{Vakulya20051967,
title = {Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts},
author = {B. Vakulya and S. Varga and A. Csámpai and T. Soós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-19544393388&doi=10.1021%2fol050431s&partnerID=40&md5=d00104af201e2c97441bfe70232cbece},
doi = {10.1021/ol050431s},
year = {2005},
date = {2005-01-01},
urldate = {2005-01-01},
journal = {Organic Letters},
volume = {7},
number = {10},
pages = {1967-1969},
abstract = {(Chemical Equation Presented) Cinchona alkaloid-derived chiral bifunctional thiourea organocatalysts were synthesized and applied in the Michael addition between nitromethane and chalcones with high ee and chemical yields. © 2005 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

2003
Z. Riedl; P. Kövér; T. Soós; G. Hajós; O. Egyed; L. Fábián; A. Messmer
Unexpected ring transformation to pyrrolo[3.2-b]pyridine Derivatives. Fused azolium salts. 22 Journal Article
In: Journal of Organic Chemistry, vol. 68, no. 14, pp. 5652-5659, 2003.
@article{Riedl20035652,
title = {Unexpected ring transformation to pyrrolo[3.2-b]pyridine Derivatives. Fused azolium salts. 22},
author = {Z. Riedl and P. Kövér and T. Soós and G. Hajós and O. Egyed and L. Fábián and A. Messmer},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-0038676260&doi=10.1021%2fjo034231a&partnerID=40&md5=b6eb268f426395d70922bf534655c8c0},
doi = {10.1021/jo034231a},
year = {2003},
date = {2003-01-01},
journal = {Journal of Organic Chemistry},
volume = {68},
number = {14},
pages = {5652-5659},
abstract = {2-Arylsulfanyl and 2-benzylsulfanylpyridinium N-arylimides (2) easily prepared from 3-aryltetrazolopyridinium salts (1) with aryl and benzylthiolates, respectively, reacted with various dipolarophiles yielding cycloadducts that underwent transformation to give tetrahydropyrrolo[3,2-b]pyridines (5, 6, and 8) in good yields. A similar rearrangement (formation of 15) was also observed in the case of parent derivatives being unsubstituted in position 2 (12). The abscence of any significant solvent effect, comparison of the sulfur and non-sulfur analogues, as well as the stereoselective nature of the observed ring transformation seem to support a sigmatropic mechanism. Structure elucidation of the products has been carried out by single-crystal X-ray diffraction and 1H NMR experiments.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2002
H. Jiao; S. Le Stang; T. Soós; R. Meier; K. Kowski; P. Rademacher; L. Jafarpour; J. -B. Hamard; S. P. Nolan; J. A. Gladysz
In: Journal of the American Chemical Society, vol. 124, no. 7, pp. 1516-1523, 2002.
@article{Jiao20021516,
title = {How to insulate a reactive site from a perfluoroalkyl group: Photoelectron spectroscopy, calorimetric, and computational studies of long-range electronic effects in fluorous phosphines P((CH2)m (CF2)7CF3)3},
author = {H. Jiao and S. Le Stang and T. Soós and R. Meier and K. Kowski and P. Rademacher and L. Jafarpour and J. -B. Hamard and S. P. Nolan and J. A. Gladysz},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-0037138658&doi=10.1021%2fja011877g&partnerID=40&md5=20aec9482e12095157aece8d3ae25c07},
doi = {10.1021/ja011877g},
year = {2002},
date = {2002-01-01},
journal = {Journal of the American Chemical Society},
volume = {124},
number = {7},
pages = {1516-1523},
abstract = {This study advances strategy and design in catalysts and reagents for fluorous and supercritical CO2 chemistry by defining the structural requirements for insulating a typical active site from a perfluoroalkyl segment. The vertical ionization potentials of the phosphines P((CH2)mRf8)3 (m = 2 (2) to 5 (5)) are measured by photoelectron spectroscopy, and the enthalpies of protonation by calorimetry (CF3SO3H, CF3C6H5). They undergo progressively more facile (energetically) ionization and protonation (P(CH2CH3)3 > 5 > 4 ≈ P(CH3)3 > 3 > 2), as expected from inductive effects. Equilibrations of trans-Rh(CO)(Cl)(L)2 complexes (L = 2, 3) establish analogous Lewis basicities. Density functional theory is used to calculate the structures, energies, ionization potentials, and gas-phase proton affinities (PA) of the model phosphines P((CH2)mCF3)3 (2′-9′). The ionization potentials of 2′-5′ are in good agreement with those of 2-5, and together with PA values and analyses of homodesmotic relationships are used to address the title question. Between 8 and 10 methylene groups are needed to effectively insulate a perfluoroalkyl segment from a phosphorus lone pair, depending upon the criterion employed. Computations also show that the first carbon of a perfluoroalkyl segment exhibits a much greater inductive effect than the second, and that ionization potentials of nonfluorinated phosphines P((CH2)mCH3)3 reach a limit at approximately nine carbons (m = 8).},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2001
T. Soós; B. L. Bennett; D. Rutherford; L. P. Barthel-Rosa; J. A. Gladysz
Synthesis, reactivity, and metal complexes of fluorous triarylphosphines of the formula P(p-C6H4(CH2)3(CF2)n -1CF3)3 (n = 6, 8, 10) Journal Article
In: Organometallics, vol. 20, no. 14, pp. 3079-3086, 2001.
@article{Soós20013079,
title = {Synthesis, reactivity, and metal complexes of fluorous triarylphosphines of the formula P(p-C6H4(CH2)3(CF2)n -1CF3)3 (n = 6, 8, 10)},
author = {T. Soós and B. L. Bennett and D. Rutherford and L. P. Barthel-Rosa and J. A. Gladysz},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-0035833299&doi=10.1021%2fom010193e&partnerID=40&md5=d9788cdddc3747bb74c47373ff998e18},
doi = {10.1021/om010193e},
year = {2001},
date = {2001-01-01},
journal = {Organometallics},
volume = {20},
number = {14},
pages = {3079-3086},
abstract = {Reactions of p-BrC6H4CH=O with Wittig reagents derived from [Ph3PCH2CH2Rfn]+ I- (Rfn = (CF2)n-1CF3; n = 6 (6a), 8 (6b), 10 (6c)) give p-BrC6H4CH=CHCH2Rfn (86-93%), which are treated with H2 and Wilkinson's catalyst to yield p-BrC6H4(CH2)3Rfn (91-94%). Reactions with n-BuLi and PCl3 (0.33 equiv) give, after workup, mixtures of the title compounds (9a-c) and the corresponding phosphine oxides (10a-c). Treatment with H2O2 gives pure 10 (a/b/c 88/57/24%), which are reduced with Cl3SiH/Et3N to 9 (a/b/c 69/82/43%). Fluorous phase affinities increase with perfluoroalkyl chain length, as quantified by CF3C6F11toluene partition coefficients (9a, 19.5:80.5; 9b, 66.6:33.4). Reaction of 9b, [Ir(COD)(μ-Cl)]2, and CO gives trans-Ir(CO)(Cl)[P(p-C6H4(CH2)3 Rf8)3]2 (76%). The IR νCO value is only slightly greater than that of Vaska's complex (1958 vs 1952 cm-1), indicating nearly negligible inductive effects of the perfluoroalkyl groups. Reaction of 9b and [Rh(COD)(μ-Cl)]2 yields Rh[P(p-C6H4(CH2)3 Rf8)3]3(Cl) (82-93%), which gives small equilibrium amounts of [Rh[P(p-C6H4(CH2)3 Rf8)3]2(μ-Cl)]2 and 9b in solution, and catalyzes the hydrogenation of alkenes under both biphasic (CF3C6F11/toluene) and monophasic (CF3C6H5) conditions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2000
L. J. Alvey; R. Meier; T. Soós; P. Bernatis; J. A. Gladysz
In: European Journal of Inorganic Chemistry, no. 9, pp. 1975-1983, 2000.
@article{Alvey20001975,
title = {Syntheses and carbonyliridium complexes of unsymmetrically substituted fluorous trialkylphosphanes: Precision tuning of electronic properties, including insulation of the perfluoroalkyl groups},
author = {L. J. Alvey and R. Meier and T. Soós and P. Bernatis and J. A. Gladysz},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-0033806523&doi=10.1002%2f1099-0682%28200009%292000%3a9%3c1975%3a%3aaid-ejic1975%3e3.0.co%3b2-m&partnerID=40&md5=2d65402388b94087e632a992ab778448},
doi = {10.1002/1099-0682(200009)2000:9<1975::aid-ejic1975>3.0.co;2-m},
year = {2000},
date = {2000-01-01},
journal = {European Journal of Inorganic Chemistry},
number = {9},
pages = {1975-1983},
abstract = {Reactions of iodides I(CH2)(m)R(f8) [m = 2-4; R(f8) = (CF2)7CF3] and LiPH2 · DME (-45 °C, THF) give the primary phosphanes PH2(CH2)(m)R(f8) (7-9; 48-76%). Radical-initiated reactions (100 °C) of 7 and H2C=CHCH2R(f8), 8 and H2C=CHR(f8) or H2C= CHCH2CH2R(f8), and 9 and H2C=CHCH2R(f8), give the title phosphanes P[(CH2)(m)R(f8)]2[(CH2)(m')R(f8)] [m/m' = 2/3 (10), 3/2 (11), 3/4 (12), 4/3 (13); 70-76%]. The symmetrically substituted phosphane P[(CH2)(m)R(f8)]3 (m = 5, 6) is similarly prepared from PH3 and H2C=CHCH2CH2CH2R(f8), analogously to previously reported homologs [m = 2 (2), 3 (4), 4 (5)]. Reactions of 10-13 and 2, 4, 5, and 6 with [Ir(COD)Cl]2 and CO give trans-Ir(CO)(Cl)(PR2R')2 (70-83%). The IR V(CO) values show a monotonic decrease with increasing numbers of CH2 groups. Phosphanes 13, 5, and 6 have the most CH2 groups, and give V(CO) values 10, 7, and 4 cm-1 higher than the unfluorinated phosphane P[(CH2)7CH3]3. Hence, 6 provides nearly complete insulation of the iridium center from the electronegative perfluoroalkyl groups. Analogous rhodium derivatives of 4 and 5 are also described.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
1999
T. Soós; G. Timári; G. Hajós
A concise synthesis of furostifoline Journal Article
In: Tetrahedron Letters, vol. 40, no. 49, pp. 8607-8609, 1999.
@article{Soós19998607,
title = {A concise synthesis of furostifoline},
author = {T. Soós and G. Timári and G. Hajós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-0033520963&doi=10.1016%2fS0040-4039%2899%2901803-1&partnerID=40&md5=81999a9a546ee5327cb16fe0787429ce},
doi = {10.1016/S0040-4039(99)01803-1},
year = {1999},
date = {1999-01-01},
journal = {Tetrahedron Letters},
volume = {40},
number = {49},
pages = {8607-8609},
abstract = {A five-step total synthesis of the furo[3,2-a]carbazole alkaloid, furostifoline, was achieved using a Pd(0)-catalyzed cross-coupling reaction.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
M. Béres; G. Hajós; Z. Riedl; T. Soós; G. Timári; A. Messmer
Valence bond isomerization of fused [1,2,3]triazolium salts with bridgehead nitrogen atom. Fused azolium salts. 19 Journal Article
In: Journal of Organic Chemistry, vol. 64, no. 15, pp. 5499-5503, 1999.
@article{Béres19995499,
title = {Valence bond isomerization of fused [1,2,3]triazolium salts with bridgehead nitrogen atom. Fused azolium salts. 19},
author = {M. Béres and G. Hajós and Z. Riedl and T. Soós and G. Timári and A. Messmer},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-0038062030&doi=10.1021%2fjo990328e&partnerID=40&md5=b757182587df1abe17b92b05593266f5},
doi = {10.1021/jo990328e},
year = {1999},
date = {1999-01-01},
journal = {Journal of Organic Chemistry},
volume = {64},
number = {15},
pages = {5499-5503},
abstract = {[1,2,3]Triazolo[1,5-a]quinolinium (7), [1,2,3]triazolo[1,5- b]isoquinolinium (8), and [1,2,3]triazolo[1,5-a]pyrazinium salts (9) when heated in trifluoroacetic acid and/or 1,2-dichlorobenzene undergo valence bond isomerization to ring-opened reactive intermediates (e.g., 8 gave 13) which can participate in (i) electrophilic substitution as nitrenium cations to yield pyrazole- and indazole-fused new heterocycles (e.g., from 13, 14, and 15 are formed), (ii) pseudoelectrocyclization (e.g., intermediate 19c leads to the pyrazolo[3,4-b]pyrazine 21), or (iii) in nucleophilic addition as carbenium cations (e.g., 1 gave the methoxy-substituted adduct 22 when heated in methanol). Comparison of these and some recent results reveals that this ring opening of fused [1,2,3]triazolium salts is a general phenomenon and is closely related to the well-known retro-electrocyclizations (called '1,5-dipolar cyclizations') of neutral fused [1,2,3]triazoles and tetrazoles.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
1997
G. Timári; T. Soós; G. Hajós
A Convenient Synthesis of Two New Indoloquinoline Alkaloids Journal Article
In: Synlett, vol. 1997, no. 9, pp. 1067-1068, 1997.
@article{Timári19971067,
title = {A Convenient Synthesis of Two New Indoloquinoline Alkaloids},
author = {G. Timári and T. Soós and G. Hajós},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-0038305874&doi=10.1055%2fs-1997-1523&partnerID=40&md5=a63d6619b19888b51f41c37c8723d3da},
doi = {10.1055/s-1997-1523},
year = {1997},
date = {1997-01-01},
journal = {Synlett},
volume = {1997},
number = {9},
pages = {1067-1068},
abstract = {A concise synthesis of two new indoloquinoline-based alkaloids (1 and 2), isolated from Cryptolepis sanguinolenta, is reported. The palladium-catalyzed cross-coupling reaction of 3-bromoquinoline derivatives with N-pivaloylamino phenylboronic acid gave the corresponding biaryls from which the indoloquinolines could be synthesized.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
T. Soós; Gy. Hajós; A. Messmer
Novel Thermal Rearrangement of Fused Diaryl-v-Triazolium Salts to Neutral Indazole Derivatives. Fused Azolium Salts Journal Article
In: Journal of Organic Chemistry, vol. 62, no. 4, pp. 1136-1138, 1997.
@article{Soós19971136,
title = {Novel Thermal Rearrangement of Fused Diaryl-v-Triazolium Salts to Neutral Indazole Derivatives. Fused Azolium Salts},
author = {T. Soós and Gy. Hajós and A. Messmer},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-0037724391&doi=10.1021%2fjo961749o&partnerID=40&md5=0c2485f0b846fc9c2940f4d85550838b},
doi = {10.1021/jo961749o},
year = {1997},
date = {1997-01-01},
journal = {Journal of Organic Chemistry},
volume = {62},
number = {4},
pages = {1136-1138},
abstract = {Alkylation of 1,3-diaryl-v-triazolo[1,5-a]benzimidazole (1) by trimethyloxonium salt afforded selectively the 4-methyl quaternary salt 2, whereas reaction with dimethyl sulfate at elevated temperature resulted in ring transformation, implying ring opening to a nitrenium cation and a subsequent electrophilic ring closure to give a benzimidazolylindazole product 5. Independent of the methylation, a similar easy thermal rearrangement was observed with alkylated 1,3-diaryl-v-triazolo[1,5-α]benzimidazolium (2 → 4) and the related benzothiazolium salt (7 → 8). Extension of the reaction to diaryl-v-triazolopyridinium salt (9 → 10 + 11) allowed observation of an additional novel reaction path that provided further support for the supposed reaction mechanism.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
1996
G. Timári; T. Soós; G. Hajós; A. Messmer; J. Nacsa; J. Molnár
Synthesis of novel ellipticine analogues and their inhibition of Moloney leukaemia reverse transcriptase Journal Article
In: Bioorganic and Medicinal Chemistry Letters, vol. 6, no. 23, pp. 2831-2836, 1996.
@article{Timári19962831,
title = {Synthesis of novel ellipticine analogues and their inhibition of Moloney leukaemia reverse transcriptase},
author = {G. Timári and T. Soós and G. Hajós and A. Messmer and J. Nacsa and J. Molnár},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-0030568131&doi=10.1016%2fS0960-894X%2896%2900521-5&partnerID=40&md5=02836787083bbbbc0a5f58ab087840d4},
doi = {10.1016/S0960-894X(96)00521-5},
year = {1996},
date = {1996-01-01},
journal = {Bioorganic and Medicinal Chemistry Letters},
volume = {6},
number = {23},
pages = {2831-2836},
abstract = {Two new ellipticine analogues were synthetized as potential non nucleoside inhibitors of reverse transcriptase and were tested Moloney leukaemia virus reverse transcriptase in vitro. Both derivatives (9a,b) showed considerable inhibitory effect; ID50 was found to be in the range of 2.8 to 4.5 x 10-5 M.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
1995
G. Halós; Z. Riedl; G. Timári; A. Kotschy; M. Béres; T. Soós; A. Messmer; S. Bátori; W. Ritzberger; J. Schantl
Fused triazoles and triazolium salts with bridgehead nitrogen atom. Novel syntheses and selective transformations Journal Article
In: Chemistry of Heterocyclic Compounds, vol. 31, no. 10, pp. 1188-1195, 1995.
Links | Tags:
@article{Halós19951188,
title = {Fused triazoles and triazolium salts with bridgehead nitrogen atom. Novel syntheses and selective transformations},
author = {G. Halós and Z. Riedl and G. Timári and A. Kotschy and M. Béres and T. Soós and A. Messmer and S. Bátori and W. Ritzberger and J. Schantl},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-34249754477&doi=10.1007%2fBF01185588&partnerID=40&md5=6f6c84b55b1d1edfbf2728071083fe25},
doi = {10.1007/BF01185588},
year = {1995},
date = {1995-01-01},
journal = {Chemistry of Heterocyclic Compounds},
volume = {31},
number = {10},
pages = {1188-1195},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
1992
G. Timári; G. Hajós; Z. Riedl; M. Béres; T. Soós; A. Messmer; S. Gronowitz
Synthesis of Polyfused Heteroaromatics by Palladium-Catalyzed Cross-Coupling Reaction Journal Article
In: Molecules, vol. 1, no. 10, pp. 236-241, 1992.
@article{Timári1992236,
title = {Synthesis of Polyfused Heteroaromatics by Palladium-Catalyzed Cross-Coupling Reaction},
author = {G. Timári and G. Hajós and Z. Riedl and M. Béres and T. Soós and A. Messmer and S. Gronowitz},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-25344479156&doi=10.1007%2fs007830050042&partnerID=40&md5=6c052680edbe88de358407e7a593d4a6},
doi = {10.1007/s007830050042},
year = {1992},
date = {1992-01-01},
journal = {Molecules},
volume = {1},
number = {10},
pages = {236-241},
abstract = {The palladium-catalyzed coupling of unsaturated halides or triflates with organostannanes (the Stille reaction) or with organo-boronic acids (the Suzuki reaction) has envolved as a powerful means of carbon-carbon bond formation. As an extention of our work on the synthesis and reactivity of positively charged bridge-head nitrogen containing fused azinium salts, we have been studying the formation of polyfused heteroaromatics with angular anellation pattern. The reaction of such positively charged salts with nucleophiles has proven to be a suitable tool for the preparation of heteroaryl dienes, which have been shown to be excellent intermediates for several ring closure reaction. A selection of results will be presented, including applications to natural product synthesis. © 1992, Springer-Verlag. All rights reserved.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

